






白井 剛
(しらい・つよし)
Tsuyoshi Shirai
略歴
- 名古屋大学大学院理学研究科生物学専攻退学
- 名古屋大学大学院工学研究科助手、生物分子工学研究所主任研究員を歴任
構造生物学研究室
卒業研究テーマ例
- ミスマッチDNA切断酵素NucSの結晶構造解析
- 糖尿病承認新薬の構造データ収集と比較解析
- コンピュータモデリングによる狂牛病プリオンのドラッグデザイン
超分子モデリングパイプラインの開発
超分子構造生物学

産業用酵素・祖先型タンパク質などの構造解析研究
- 研究の応用領域
- タンパク質工学・ドラッグデザイン・データベース・情報生物学解析システム開発
- 産官学連携で求めるパートナー
- 酵素・タンパク質関連企業
医薬品関連企業
IT関連企業
We are developing the methods in analyzing protein/proteome system by combining the bioinformatics and the experimental, mainly X-ray crystallography, approaches.
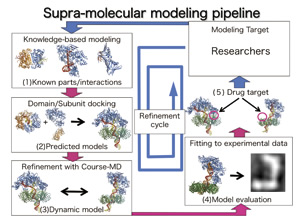
Development of Supra-molecular Modeling Pipeline
The determined structures of proteins in the Protein Data Bank have been conventionally classified manually by expert researchers. In order to cope with the rapidly inflating database, the fully-automated 2ry PDB processing system (SIRD) has been developed through our research (http://sird.nagahama-i-bio.ac.jp/sird/). This system can be used to predict unknown structures of biological supra-molecules (large functional assemble of bio-molecules in organisms). Under a collaboration with my colleagues, the SIRD system is further developed into supra-molecular modeling pipeline, which will be the first integrated computational resource for modeling biological supra-molecules and assistingstructurebased drug designs targeted to macromolecular interactions, with the support from Platform for Drug design, Informatics, and Structural lifescience (PDIS)project.
Computer-aided modeling of supramolecular complexes
By using SIRD and other developed methods, practical modeling of protein complexes and supra-molecules has been executed, mainly under collaboration with researchers in this and other institutes. For example, the computational models for prion fiber (responsible factor of BSE), parts of replisome (dynamic supra-molecular complex required for DNA replication), and complexes of carbohydrate metabolizing enzymes have been constructed, and used for predicting functions or interactions, and for designing experiments.
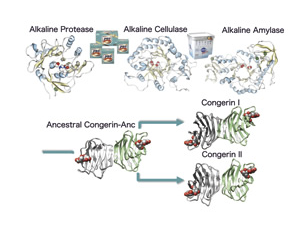
Structural analyses of industrial enzymes, and ancestral proteins
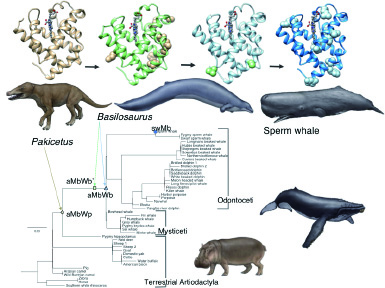
The enzymes that are resistant to extreme conditions such as high-alkaline or high-temperature, are industrially used as the builder of detergents. The structures of detergent builder protease, cellulase, and amylase were determined with X-ray crystallography, and the molecular mechanisms for alkaline-adaptation of proteins was proposed. Also the glucosidase from the deepest-sea bacteria was analyzed to understand protein adaptation to a high-pressure. The bioinformatics techniques can be used to calculate ancient structure of genes/proteins. The ancestral sequence of fish congerin (innate-immune protein) was predicted by using computer. The ancestral congeringene was synthesized, and its three-dimensional structure was determined as the first time for a full-length ancestral protein.
Hijikata A, Tsuji T, Shionyu M, Shirai T, Decoding disease-causing mechanisms of missense mutations from supramolecular structures, Sci. Rep. 7, 8541 (2017)
Shionyu-Mitsuyama C, Hijikata A, Tsuji T, Shirai T, Classification of ligand molecules in PDB with graph match-based structural superposition. J. Struct. Funct. Gen., 17, 135-146 (2016)
Nakae S, Hijikata A, Tsuji T, Yonezawa K, Kouyama K, Mayanagi K, Ishino S, Ishino Y, Shirai T, Structure of EndoMS-DNA complex as mismatch-restriction endonuclease. Structure, 24, 1960-1971 (2016)
Tsuji T, Yoda T, Shirai T, Deciphering Supramolecular Structures with Protein-Protein Interaction Network Modeling. Sci. Rep. 5, 16341 (2015)
Shirai T, Saito M, Kobayashi A, Asano M, Hizume M, Ikeda S, Teruya K, Morita M, Kitamoto T, Evaluating prion models on comprehensive mutation data of mouse PrP, Structure, 22, 560-571 (2014)