






山本 哲志
(やまもと・てつし)
Tetsushi Yamamoto
略歴
- 大阪医療技術学園専門学校 臨床検査技師科卒業
- 三田市民病院 臨床検査科、神戸大学医学部附属病院 検査部を経て本学へ
機能診断学研究室
DMDの心筋症は成人の心筋症と同様に内服加療を行うことで生命予後が改善することが知られているが、その発症年齢は様々であり一部の症例は左室の収縮障害すら生じない(Figure1)ことが判明した。DMDの心筋症は左室後壁の収縮障害として現れ、次第に左室全体に収縮低下が広がり、さらに左室拡大を呈し拡張型心筋症様の変化を全症例がきたすとされていたが、適切な内服加療を行うと左室拡大を生じる症例は全体の2割程度(Figure2)である。しかし、多くの患者が左室の拡大を呈さないにも関わらず死因の半数を心筋症の発症が占めており、DMD患者の心筋症の発症予測や評価、進行のマーカーとして心電図検査結果や心臓超音波検査結果の解析が不十分であるといえる。
そこで本研究室ではDMDを中心にベッカー型筋ジストロフィー(BMD)などの類似の筋ジストロフィー症も含め以下の研究を通じて、現行の治療における予後の予測、疾患の治療の標的分子を明らかにし新たな治療法開発につながる知見を得ること、その治療効果判定の方法などを見出すことを目指す。
ジストロフィン遺伝子変異と心機能の関連
DMDの心筋症発症を予測
DMDの呼吸機能について


- 研究の応用領域
- 心不全の治療薬・診断薬の開発、筋ジストロフィーの遺伝子治療
- 産官学連携で求めるパートナー
- 医療機関、治療薬・診断薬関連企業
DMD (Duchenne muscular dystrophy) is the most common inherited muscle disease appearing in childhood, affecting one of 5000 to 10000 male newborns. DMD is caused by mutations in the DMD gene, located on the X chromosome, and is characterized by complete loss of muscle dystrophin, a protein that links the cytoskeleton to the extracellular matrix to form the dystrophin glycoprotein complex. Male children affected by DMD typically show initial muscle weakness at the age of 3 to 5 years, with weakness progressing with age, and eventually resulting in loss of ambulation by the age of 12 years. Early death of patients with DMD is caused by wasting of respiratory or cardiac muscles. Recently, the life expectancy has increased, from 15 to 19 to >30 years, through the benefits of developments in respiratory care such as NIPPV. This increase in lifespan has allowed cardiomyopathy to emerge as a major cause of morbidity and mortality in patients with DMD.
It is known that cardiomyopathy in patients with DMD first manifests as left ventricular (LV) systolic dysfunction of the posterior LV wall, which gradually becomes LV-wide systolic dysfunction, followed by LV dilation and dilated cardiomyopathy-like changes, and that medical treatment for adult cardiomyopathy improves prognosis of cardiomyopathy in DMD. Our study however, revealed that the onset age of LV systolic dysfunction in DMD varies and some patients do not even develop LV systolic dysfunction (Figure 1). Further, in case of appropriate medical therapy, LV dilation occurs in only about 20% of patients (Figure 2). Thus, the development of cardiomyopathy accounts for half of all deaths in DMD patients, even though many patients do not present with LV dilation, suggesting that the results of electrocardiography and echocardiography are inadequately analyzed as markers for the prediction, evaluation, and progression of cardiomyopathy in DMD patients.
In functional diagnostics laboratory, our aim is as follows: to predict the prognosis of therapies, to identify target molecules for the treatment of the diseases, to gain knowledge that will lead to the development of new therapies, and to find methods to determine the effectiveness of the therapies for muscular dystrophy diseases such as DMD and Becker’s muscular dystrophy.
Relation between mutations in the DMD gene and cardiac function
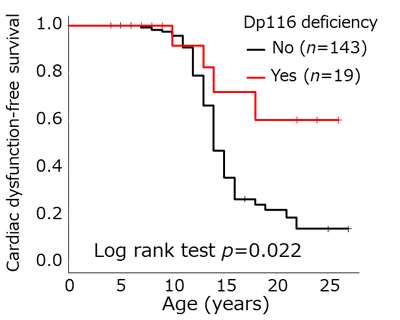
DMD is the largest human gene, consisting of 79 exons. Four internal promoters, located in introns 29, 44, 55, and 62, encode the short isoforms Dp260, Dp140, Dp116, and Dp71, respectively. We have previously shown that the onset of cardiomyopathy is significantly delayed in patients with Dp116 deficiency than in those with Dp116 (Figure 3). We will further investigate the relationship between genetic abnormalities and cardiac function by examining other isoforms and combinations of mutation types.
Predicting the onset of cardiomyopathy in DMD
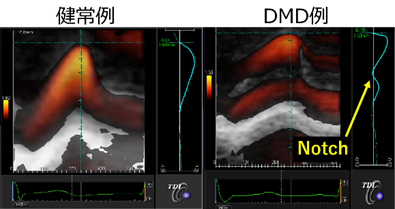
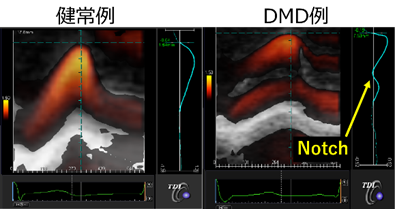
Electrocardiography and echocardiography are the most common methods of evaluating cardiomyopathy in DMD. Many patients with DMD show electrocardiography abnormalities such as abnormal Q waves and ST-T changes even before onset of cardiomyopathy diagnosed by echocardiography. However, DMD patients do not present with myocardial infarction as suggested by electrocardiography abnormalities. There are no detailed reports on the relationship between measurements such as QRS amplitude and cardiac function. In other words, despite the fact that electrocardiography testing can be easily performed in many facilities, the meaning of electrocardiography abnormalities remains unclear. In the echocardiography, we have reported that transmural myocardial strain profile, which is a type of strain method that can detect a subtle LV systolic dysfunction, shows an abnormal notch in patients with LV normal contraction by conventional echocardiography (Figure4). However, the mechanism of LV systolic dysfunction and dilation has not yet been clarified, and much remains unknown. Therefore, using the electrocardiography and the echocardiography in DMD, we will establish a new evaluation method for cardiomyopathy in DMD, and develop knowledge for evaluation markers for therapeutic medication.
Respiratory function in DMD
Although non-invasive respiratory management such as NIPPV has reduced respiratory-related deaths in DMD to about half, no report has examined respiratory function in DMD, and many things remain unknown. Therefore, we will focus on the basics of the disease, and clarify the relationship between changes over time, cardiac function, and genetic abnormalities, including isoforms and mutation types.
Nagai M, Awano H, Yamamoto T, Bo R, Matsuo M, Iijima K. The ACTN3 577XX Null Genotype Is Associated with Low Left Ventricular Dilation-Free Survival Rate in Patients with Duchenne Muscular Dystrophy. J Card Fail. 2020 Oct;26(10):841-848. doi: 10.1016/j.cardfail.2020.08.002. Epub 2020 Aug 10.
Rani AQM, Yamamoto T, Kawaguchi T, Maeta K, Awano H, Nishio H, Matsuo M. Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients. Int J Mol Sci. 2020 May 18;21(10):3555.
Yamamoto T, Awano H, Zhang Z, Sakuma M, Kitaaki S, Matsumoto M, Nagai M, Sato I, Imanishi T, Hayashi N, Matsuo M, Iijima K, Saegusa J. Cardiac Dysfunction in Duchenne Muscular Dystrophy Is Less Frequent in Patients With Mutations in the Dystrophin Dp116 Coding Region Than in Other Regions. Circ Genom Precis Med.2018 Jan;11(1):e001782. doi: 10.1161/CIRCGEN.117.001782.
Yamamoto T, Taniguchi-Ikeda M, Awano H, Matsumoto M, Lee T, Harada R, Imanishi T, Hayashi N, Sakai Y, Morioka I, Takeshima Y, Iijima K, Saegusa J, Toda T. Cardiac involvement in Fukuyama muscular dystrophy is less severe than in Duchenne muscular dystrophy. Brain Dev. 2017 Nov;39(10):861-868. doi: 10.1016/j.braindev.2017.05.008.
Yamamoto T, Tanaka H, Kurimoto C, Imanishi T, Hayashi N, Saegusa J, Morinobu A, Hirata KI, Kawano S. Very early stage left ventricular endocardial dysfunction of patients with hypereosinophilic syndrome. Int J Cardiovasc Imaging. 2016 Sep;32(9):1357-1361. doi: 10.1007/s10554-016-0917-x.