






依田 隆夫
(よだ・たかお)
Takao Yoda
略歴
- 東京大学大学院理学系研究科博士課程修了
- 岡崎国立共同研究機構・分子科学研究所研究員、日本学術振興会リサーチアソシエートを経て本学へ
計算構造生物学研究室
卒業研究テーマ例
- 分子動力学シミュレーションによる、蛋白質フォールディングの研究
- 分子動力学シミュレーションによる、抗菌ペプチドの立体構造と作用機構に関する研究
- 分子シミュレーションによる、タンパク質分子複合体形成に関する研究
- 分子動力学シミュレーションによる天然変性蛋白質の構造の研究
拡張アンサンブル法による蛋白質の折れ畳みシミュレーション
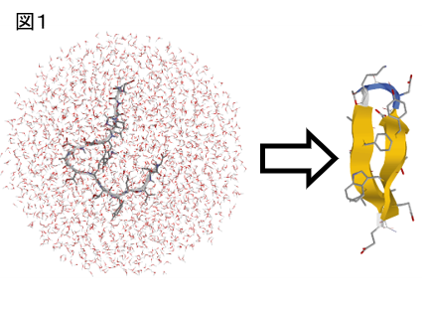
我々は分子動力学法(MD)などのシミュレーション技法を活用して、タンパク質やペプチド分子の自発的な構造形成機構を研究してきた。水溶媒中にランダムな構造の蛋白質を配置(図1の左半分)し、蛋白質分子が折れ畳むまでシミュレーションを行う。これを効率化するために拡張アンサンブル法という手法を適用している。
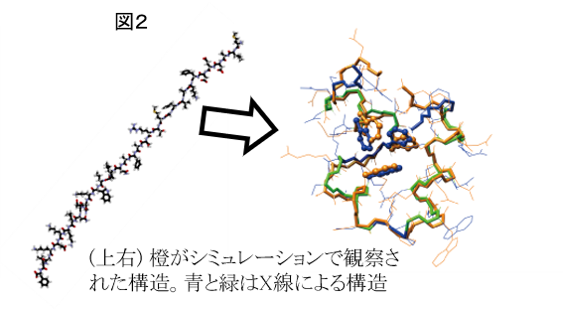
我々は同種法を16残基のβ - ヘアピンペプチド(図1)や36残基からなる小蛋白質(図2)に適用した。いずれにおいても天然構造に非常に近い構造への折れ畳みが観察された。各図の右半分に陽溶媒中でのシミュレーションで実際に観察された立体構造を示した。このように、ペプチド分子や小蛋白質の折れ畳み構造を陽溶媒中でのシミュレーションによって出現させられるようになってきた。
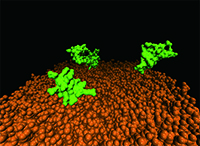
分子シミュレーションによる抗菌ペプチドの機能と構造揺らぎの研究
- 研究の応用領域
- 準安定構造を考慮した機能性分子設計
ペプチド分子の構造機能相関 - 産官学連携で求めるパートナー
- 当該応用分野の研究を行っている方
Proteins are macromolecules that play important roles in most physiological processes. We should be able to understand the molecular mechanisms of the efficiency and the specificity of proteins’ functions based on their structures and dynamics. We are studying protein folding and function by use of techniques of molecular dynamics (MD) simulation. MD is useful for observing and studying protein’s conformational changes and fluctuations at atomic level. However, MD of protein systems has some limitations due to its computational cost. Thus methods for efficient simulations are required.
We adopt replica-exchange method (REM) and its derivatives, which were introduced by Sugita and Okamoto for protein systems at first, for efficient simulations. Our research activity involves software development and its application to biological molecules.
Protein Folding Studies by Molecular Simulations
We performed a folding simulation of a 16-residueβ-hairpin peptide that is called “G-peptide”, started from the fully extended conformation. We used MUltiCAnonical Replica-Exchange Method (MUCAREM) for efficient sampling. We successfully observed the folding events to its native conformation, and studied the folding scenario of G-peptide. We found that the turn formation is a key step of the folding of the hairpin peptide, and this prevents misfolding to non-native conformations with wrong secondary structures. From the analyses of the simulation data, it was also shown that side-chain hydrogen bonds near the turn region are formed in an early stage of folding.
We also applied the method to a 36-residue globular protein that is called “HP36”. This simulation was also started from the fully extended conformation of the protein molecule. Again, we successfully observed folding events to its native structure, during the production simulation shorter than the experimental folding time. We found again that a turn formation is important for folding of HP36 and that packing and dehydration of hydrophobic-core side chains take place in a later stage of folding.
Simulation Study of Antimicrobial Peptide
We are now studying functions of defensin, utilizing the methodologies that we applied to protein folding studies. There are three families of defensin (α, β, and θ), out of which α and β have a well-characterized fold stabilized by disulfide bridges. They act as anti-microbial agents in the small intestine, the epithelium, and neutrophils. Defensins are known to cause leakage of bacterial cells, however, its molecular mechanism is not well known. Aims of our current studies are to simulate the interaction between defensin and membrane and to explore the fluctuated conformations of the peptide in solution. Our recent simulation results have suggested the underlying mechanism of some defensins’ activity changes by mutations.
T.Yoda, Y.Sugita, & Y.Okamoto, ‘Salt ef f ects on hydrophobic-core formation in folding of a helical mini protein studied by molecular dynamics simulations’, Proteins 82 (6), 933-943 (2014).
依田、杉田、岡本「疎水コアとα-ヘリックスを含む小蛋白質のフォールディングシミュレーション」, 生物物理(日本生物物理学会誌), 52 (1), 022-023 (2012)
T.Yoda, Y.Sugita, & Y.Okamoto, ‘Hydrophobic core formation and dehydration in protein folding studied by generalized-ensemble simulations’, Biophys. J. 99 (5), 1637-1644 (2010)
T.Yoda, Y.Sugita, and Y.Okamoto, Cooperative folding mechanism of a β-hairpin peptide studied by a multicanonical replica-exchange molecular dynamics simulation, Proteins 66, 846-859 (2007)
T.Yoda, Y. Sugita and Y.Okamoto, Comparisons of force f i elds for proteins by generalized-ensemble simulations, Chem. Phys. Lett. 386, 460-467 (2004)