






高宮 脩
(たかみや・おさむ)
Osamu Takamiya
略歴
- 大阪工業大学工学部応用化学科卒業、奈良県立医科大学小児科学専修生学位取得退学
- 信州大学医学部保健学科教授、同大学院医学系研究科教授、同大学名誉教授
血液凝固第VII 因子欠乏・異常症の分子病態の解析
血液凝固診断試薬の評価と開発
血栓性疾患の予防のため投与されるwarfarinのモニタリングに使われるOwren-typeの試薬はウシ脳抽出物からのトロンボプラスチンが用いられてきたが、狂牛病のためウシ脳を用いることが出来なくなった。カイコを用いて遺伝子組み換え型ウシ組織因子を発現させた新規試薬を作成して、有用性のあることを報告した。これまで培ってきた臨床検査経験や知識、情報に基づき新規の凝固・線溶試薬の開発を行いたい。
- 研究の応用領域
- 変異第VII 因子の分子病態、診断試薬の開発・改良
- 産官学連携で求めるパートナー
- 医療機関、診断試薬関連企業
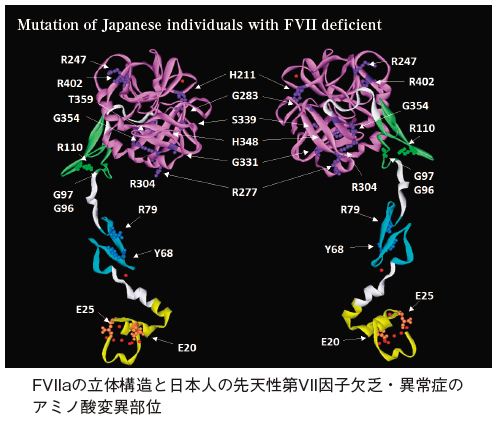
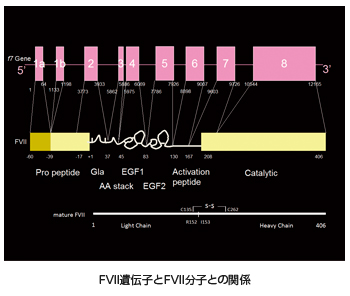
Factor VII (FVII) participates in the initial stages of blood coagulation. It is a vitamin K-dependent glycoprotein that is synthesized in the liver, secreted into the blood and circulates as a single-chain zymogen composed of 406 amino acid residues with an approximate molecular weight of 50 kDa. The light chain consists of an aminoterminal γ-carboxyglutamic acid rich domain, followed by two EGFlike domains. The heavy chain consists of the serine protease catalytic domain. The gene for FVII is located on chromosome 13 at q34-qter, spans 12.8 kilobase pairs, and contains exons 1a and 1b (which encode the 5′ untranslated region and most of the pre-pro leader sequence) and exons 2 to 8 (which encode the mature protein).
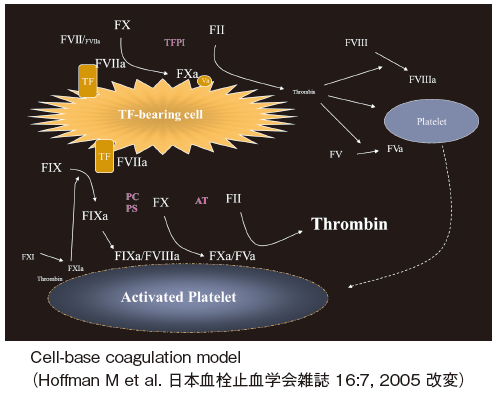
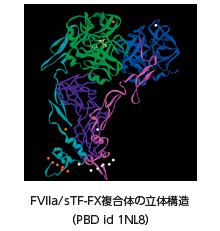
FVII and FVIIa form a one-to-one stoichiometric complex with tissue factor TF on endothelium cells recruited by vessel damage. Once complexed to TF, FVII zymogen is converted to its active form by various coagulation proteases such as FXa. The TF-FVIIa complex readily activates zymogen FX and FIX to active enzymes and promotes fibrin-formation.
Hereditary FVII deficiency is a rare autosomal recessive bleeding disorder that has an estimated prevalence of one per 500,000 in the general population.
FVII knockout mice die at birth, or shortly after, from major abdominal and intracranial hemorrhaging. In humans, the clinical phenotype exhibited by patients with FVII deficiency correlates with poor diagnostic laboratory tests.
Differences between the clinical and laboratory phenotypes may be due to the complexity of biochemical interactions that occur with the initiation of hemostasis or because of a lack of sensitivity and accuracy in the assays used to measure very low levels of FVII:C.
We have reported various problematic issues with currently available commercial reagents and/or reference standard plasma.
However, no data regarding the inter-laboratory variability of FVII:C determinations in samples with low to very low FVII:C have so far been reported. It is very important to compare the results of different determinations of FVII deficiency for accurate diagnosis and treatment. We distributed three deficient plasma samples prepared via immunoaffinity chromatography among 58 laboratories in Japan. These samples were measured using the routine protocol in place at each laboratory and a calibration reference plasma and recombinant thromboplastin were supplied as common reagents. We conclude that the results of the FVII:C assays, especially for cases with very low to low FVII:C concentrations, are greatly influenced by the number of plasma dilutions used in constructing a standard activity curve in additional to the type of thromboplastin and calibration plasma utilized to perform the tests. These results should prove useful in the development of more sensitive and accurate systems to measure FVII:C for the diagnosis and therapy of FVII deficiency.
Properties of a recombinant bovine tissue factor expressed by Silkworm pupae and its performance as an Owren-type prothrombin time reagent for warfarin monitoring. Thrombosis Research. 130:520-527. 2012 Okuda M, Taniguchi T, Takamiya O.
Factor VII def iciency due to compound heterozygosity for Lue-48Pro mutation and a novel Pro260Leu mutation. Clinical and Applied Thrombosis and Haemostasis 17:E205-210 2011 Kogiso N, Taki M, Takamiya O
Japanese collaborative study to assess inter-laboratory variation in factor VII activity assays. Journal of Thrombosis Haemostasis 5: 1686-1692, 2007 Takamiya O,Ishikawa S, Ohnuma O, Suehisa H, Iijima K, Kayamori Y, Bando S, Higashi K
Molecular mechanism of dysfunctional factor VII associated with the homozygous missense mutation 331Gly to Ser. Thrombosis Haemostasis 93: 414- 419, 2005. Takamiya O, Kimura S
A patient homozygous for a Gly345Cys mutation in factor VII that results in severely impaired secretion of the molecule, but not complete def iciency. British Journal of Haematology 124:336-342, 2004. Takamiya O, Hino K