





フロンティアバイオサイエンス学科
 高橋 健一(たかはし・けんいち)
高橋 健一(たかはし・けんいち)
Ken-ichi Takahashi
専門分野/生物物理学、計算構造生物学
研究キーワード/タンパク質、RNA、立体構造、データベース解析、分子シミュレーション
職位:准教授
学位:博士(理学)(名古屋大学)
- 名古屋大学大学院理学研究科博士課程修了
- 名古屋大学大学院理学研究科助手を経て本学へ
研究テーマ
生命現象をつかさどる主要な分子である、蛋白質やRNAを深く理解することで生命の基本的なしくみを理解したいと考えている。特にタンパク質やRNAの機能発現の立体構造的基盤、その立体構造の構築原理、および、構造や機能の進化の過程を理解することを目指して、構造データベースの統計解析、分子モデリング、分子シミュレーションなどを行っている。またそれらを基にして、生体分子の構造・機能の予測や新規の有用な分子の設計などのツールの発展を目指す。
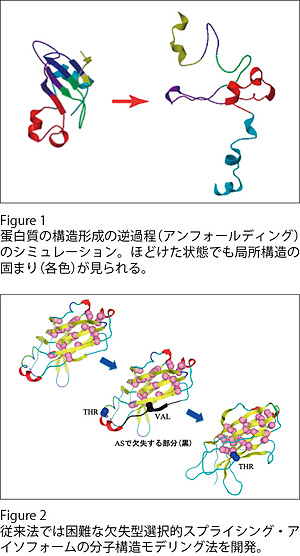
(1)タンパク質のフォールディング過程のシミュレーション
タンパク質がほどけた状態から天然構造へ自然に構造形成される分子機構を、その逆過程であるアンフォールディング過程のシミュレーションを実行することにより研究してきた。アンフォールディングが進み、全体的なコンフォメーションが大きく変化し続ける段階に達しても、ポリペプチド鎖の局所的な部分がそれぞれ比較的コンパクトな構造をとる傾向が見られた。それら局所的部分は、モジュールと言う天然構造におけるコンパクトな部分構造に対応していたため、タンパク質が階層的に折れ畳まれていく機構が示唆された(Figure 1)。この知見を取り込むことでフォールディング過程のシミュレーションを加速し、現実的な時間で天然構造に到達させることを目指す。
(2)選択的スプライシング・アイソフォームの分子モデリング法の開発
選択的スプライシング(AS)は、真核生物の複雑な分子システムを成り立たせるために、タンパク質の多様性を増加させる主な要因の1つと考えられている。しかし、そのように産生されるタンパク質(ASアイソフォーム)の構造や機能はよくわかっていない場合がほとんどである。そこで、AS の効果がタンパク質産物の構造や機能にどう及ぶかを推定し、ASアイソフォームの実験的ターゲットを提案するために、ASアイソフォームのための新しい分子モデリング法を開発している。特に構造のコア領域をコードするエクソンが欠失することで、欠失のない鋳型構造から大きな構造変化が予想される場合に適用できる方法を目指して開発している(Figure 2)。
(3)生体分子の相互作用と構造変化の解析
変異体の表現型からタンパク質の働くしくみに迫るとき、変異によりどう構造や相互作用などが変化するかが重要な問いである。その中で、ヘリックスの中にプロリンへの変異がおきると、どの程度ヘリックスが変形し、どう相互作用に影響を与えるのか、その推定法の作成を目指し、立体構造データベースからプロリンを含むヘリックスを集め統計解析を行っている。また変異の影響を調べる方法として分子シミュレーションの性能を調べている。
| 研究の応用領域 | 産官学連携で求めるパートナー |
|---|---|
| タンパク質・ゲノムの機能解析、タンパク質の設計 | 選択的スプライシングの関わる現象や疾病を研究対象とする企業、大学、国・地方自治体の研究機関 |
Topics of research
Protein and RNA are key molecular machines producing various biological phenomena. Through deep understanding of the physicochemical behavior of protein and RNA, fundamental mechanism of life must be clarified. In particular, toward understanding structural bases on which their functions are expressed, principles of self-organization of their native structures, and evolutionary processes producing such structures and functions, we have performed statistical analyses of structural data, molecular modeling and molecular simulation. Based on these studies, bioinformatics tools such as those for predicting structure and function of uncharacterized genes' products and designing new useful molecules, will be also improved.
(1) Molecular dynamics simulation of protein folding
Molecular mechanisms of self-organization to ordered native protein structures from disordered conformations have been studied by computer simulation of protein unfolding, a reverse process of the structural organization of proteins. In a stage of the unfolding process where a protein was extensively unfolded and kept on dynamically changing its overall conformation, we have found that local parts of the polypeptide chain tend to take relatively compact conformations. These parts correspond to compact substructures, "modules", in the folded structure of the protein, which suggests a hierarchical process of protein folding (Figure 1). By utilizing this finding, we aim at accelerating in silico protein folding to find ordered native structures in practical time.
(2) Development of molecular modeling method of alternative splicing isoforms
Alternative splicing (AS) is thought to be one of the major processes that increase the variation of product proteins sufficient to build complex molecular systems of eukaryotes. Structures and functions of most AS isoforms, however, remain to be elucidated. For inferring effects of AS on structures and functions of protein products and proposing experimental targets of AS isoforms, we have been developing a new strategy for molecular modeling of alternative splicing isoforms, especially for cases where large structural differences from template structures are anticipated because of the deletion of exons encoding parts of structural core regions (Figure 2).
(3) Analysis of interaction and structural change of biomolecules
In approaching the subject of protein operating principle from mutant phenotype data, it is important to ask how the mutations affect protein structure and inter-molecular interaction. Specifically, how do mutations to proline within helices bend the helical structures and affect the binding surface to other molecules? Aiming to develop an estimation method for such phenomena, we have undertaken statistical analyses of proline-containing helices collected from the protein structure database, and also evaluated the reliability of molecular simulation as a method to investigate structural effects of mutations.
主な業績論文等
- Shionyu M., Takahashi K. & Go M. AS-EAST: a functional annotation tool for putative proteins encoded by alternatively spliced transcripts. Bioinformatics 28:2076-2077 (2012)
- Shionyu M., Yamaguchi A., Shinoda K., Takahashi K. & Go M. AS-ALPS: a database for analyzing the effects of alternative splicing on protein structure, interaction and network in human and mouse. Nucleic Acids Res. 37:D305-309 (2009)
- Shinoda K., Takahashi K. & Go M. Retention of local conformational compactness in unfolding of barnase: contribution of end-to-end interactions within quasi-modules. BIOPHYSICS 3:1-12 (2007)
- Takahashi K., Baba S., Koyanagi Y., Yamamoto N., Takaku H. & Kawai G. Two basic regions of NCp7 are sufficient for conformational conversion of HIV-1 dimerization initiation site from kissing-loop dimer to extended-duplex dimer. J. Biol. Chem. 276:31274-31278 (2001)
- Takahashi K., Noguti T., Hojo H., Ohkubo T. & Go M. Conformational characterization of designed minibarnase. Biopolymers 58:260-267 (2001)